

导语:“冰桶挑战”从美国一路风靡到了中国,该活动发起人旨让人们在冰桶从头淋下造成身体麻木状态而感受“肌肉萎缩性侧索硬化症”患者的生存状况,希望更多人知道被称为渐冻人的罕见疾病,同时也达到募款帮助治疗的目的。随着冰桶挑战的不断升级,越来越多的目光投给了“罕见病群体”,盘点我们所不曾了解的“罕见病”,让我们投以更多持续更多的关爱。
“渐冻人”——肌肉萎缩性侧索硬化症

斯蒂芬·威廉·霍金
“肌肉萎缩性侧索硬化症”是一组运动神经元疾病的俗称,该病因为患者大脑、脑干和脊髓中运动神经细胞受到侵袭,患者肌肉逐渐萎缩和无力,以至瘫痪,身体如同被逐渐冻住一样,故俗称“渐冻人”。像人们熟知的一代理论物理学大师、科学巨匠霍金就是位“渐冻人”。

渐冻人需要承受大脑清醒却无法支配自己人生的巨大痛苦。目前,医学界并无特效药物治疗“渐冻人”。大多患者主要依靠进口价格昂贵又未纳入医保的进口药利鲁唑来延缓病情。使用此药每月大致需要花费5000元,国产药每月需要1200元左右,这一花费尚未计入出现肺部感染时需要购入呼吸机的费用,以及对“渐冻人”护理的费用。
而对病人家属而言,“病人的生命延续时间,很大程度上依赖‘渐冻’期间的护理条件。”平时加强病人的营养摄入、日常护理,以及注重病友和家属的精神心理状态调整,是治疗“渐冻人”的一大关键。除了昂贵的治疗费用,病人家属仍需在生理、心理方面进行极大的投入。
“瓷娃娃”——成骨不全症

瓷娃娃病即成骨不全症,又称脆骨症、原发性骨脆症及骨膜发育不良等。其特征为骨质脆弱、蓝巩膜、耳聋、关节松弛,是一种由于间充质组织发育不全,胶原形成障碍而造成的先天性遗传性疼痛,是一种先天性骨胳病。其特征为骨质脆弱,容易骨折。打个喷嚏、提个被子,甚至一个拥抱,都可能让他们骨折。成骨不全症的典型症状之一是蓝色巩膜,其患者又被称之为“蓝精灵”

该病症轻者可无症状,正常身高,通常寿限,仅轻度易发生骨折。重者残废,甚至造成死亡。一般出现的症状为骨脆性增加,轻微损伤即可引起骨折,常表现为自发性骨折,或反复多发骨折。可有脊柱侧凸,骨盆扁平,或有身材矮小。蓝巩膜、巩膜变薄,透明度增加。进行性耳聋源自听骨硬化、声音传导障碍或有人认为是听神经出颅底时被卡压所致。牙齿发育不良,灰黄,切齿变薄,切缘有缺损,关节松弛,肌腱及韧带的胶原组织发育障碍可有畸形,关节不稳定。
目前对于成骨不全暂无特殊治疗。唯一能做的就是尽量预防骨折,要严格的保护患儿,一直到骨折趋势减少为止,但又要防止长期卧床的并发症。对骨折的治疗同正常人。由于骨折愈合较迅速,固定期可短。畸形严重者可采取措施矫正畸形,改善负重力线。
“蜘蛛人症”——马凡综合征
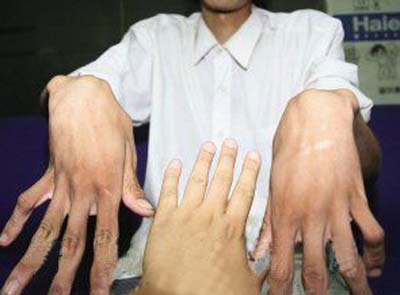
蜘蛛人指样
小儿科中有一群这样的患者,大部分高高瘦瘦,手臂极长,手指也很长,别的孩子上半身和下半身基本一样长,可他的下半身很明显比上半身长一截从其外观即可看出是马凡综合征。因身体结缔组的异常,常造成特殊的蜘蛛状指、眼睛晶状体脱位、身长高而细且关节过度伸展造成脊椎织侧弯,并常合并二尖瓣脱垂主动脉根部瘤状样扩大,也常合并下颔颜面骨发育不全,造睡眠障碍,因此又称为指趾过长症候群。
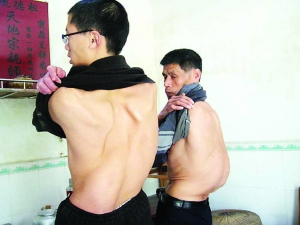
马凡氏综合征病症主要表现在骨骼、眼及心血管系统,凶险之处是先天不足造成主动脉等扩张,直至导致主动脉大破裂猝死。
2001年新年刚过的第3天,中国男排四川籍名将主力副攻手身高2米07的朱刚在训练中突感剧烈胸痛晕倒训练场,当即被送入当地医院,经过12小时的手术抢救却仍然未能挽留住这位国手的生命,年仅30岁。而更早的在上世纪80年代中期前后,中国男篮曾拥有一位身高超过2米,身材高挑匀称弹跳极佳运动灵活,被认为亚洲球员里姚明之前曾有过的具极大潜力的国家队年轻中锋运动员韩鹏山在火车上正取行李要下车时,突发剧烈胸痛,当即晕倒很快心跳呼吸停止,其猝死之快而根本未能有机会救治。事后解剖证实朱刚和韩鹏山都是猝死于“马凡氏综合症”的严重并发症——胸主动脉夹层动脉瘤破裂。
“月亮孩子”——白化病

白化病是由于酪氨酸酶缺乏或功能减退引起的一种皮肤及附属器官黑色素缺乏或合成障碍所导致的遗传性白斑病。白化病患者由于酪氨酸酶缺乏或功能减退,患者全身黑色素缺失。他们拥有月光一样白色的头发和皮肤,极易被阳光晒伤,温暖的阳光反而成为禁忌,被称为“月亮孩子”。

非洲的白化病“黑人”
目前药物治疗无效,仅能通过物理方法,尽量减少紫外辐射对眼睛和皮肤的损害。还可以使用光敏性药物、激素等治疗后使白斑减弱甚至消失。此外还需关注白化病患者心理方面的问题。白化病除对症治疗外,目前尚无根治办法,应以预防为主,通过遗传咨询禁止近亲结婚,同时进行产前基因诊断也可预防此病患儿出生。
据统计,非洲曾有多名白化病人惨遭杀害并肢解,他们大部分都是儿童。黑人白化病患者在这些贫穷落后的国家被看作为异类,命运极其悲惨。一些巫师会将他们的皮肤、骨骼以及内脏当成宝物、商人们会收集白化病人的身体器官作为求财法物、大维多利亚湖上的渔民更是相信用这些患者的头发结成的网能够捕到更多的鱼。因此这些不幸的白化病患者就像野生动物一样被人猎杀。
吸血鬼症——卟啉症
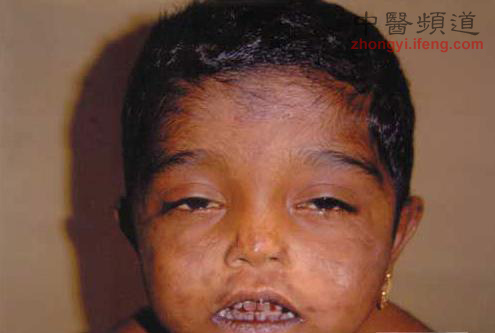
卟啉症又名血紫质病,是血红素合成途径当中,由于缺乏某种酶或酶活性降低,而引起的一组卟啉代谢障碍性疾病。主要临床症状包括光敏感、消化系统症状和精神神经症状。卟啉症有众多表现形式,比较常见的一种是急性间歇型卟啉症(AIP),英国的“疯子国王”乔治三世就是这种疾病的受害者之一。最严重的卟啉症是先天红血球生成卟啉症(CEP),患者需要输血和血红素进行缓解、畏惧阳光、面色苍白的悲惨命运被怀疑是吸血鬼故事的起源。

在艾西恩的《收割》中有这样的描述“卟啉症,一种十分罕见的基因突变病症,至今无法治愈。患者缺乏造血功能,不得不依靠食取动物的内脏来获取生存所必需的养分。其中的一些人甚至会去猎食人类而成为可怕的罪犯。在现实中,他们是数量微乎其微的最接近吸血鬼的生物。令人惊讶的是,这种疾病似乎是在成年以后才突然形成的。”
实际上,卟啉症患者的寿命通常都非常之短。紫质的代谢物是一种对光很敏感的物质,部份种类的卟啉症可能会因为这种代谢物的影响而使患者的皮肤对阳光敏感。这类患者若曝晒过量阳光,皮肤就会起水泡、溃烂,最严重时甚至能造成死亡。所以有这种症状的患者为了尽量避免接触阳光与紫外线,外出时会用黑布大面积罩住头、手等身体暴露在外的部份,在室内时也会尽量降低光量。患上吸血鬼症的人,身具红色的骨骼和尿液,黑褐色的牙齿,只能生活在黑暗中,需要补充血液增加血色素。
蝴蝶宝贝症——大疱性表皮松懈症
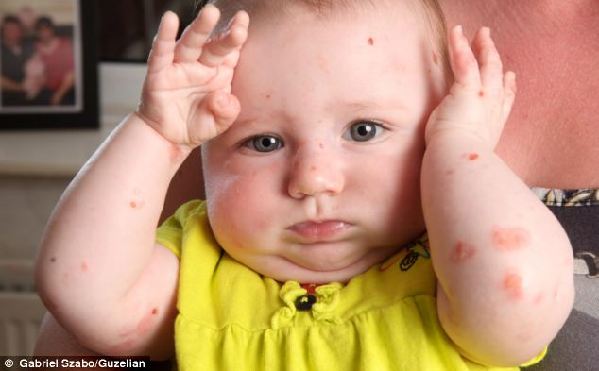
皮肤在受到轻微摩擦或碰撞后出现水疱及血疱
大疱性表皮松解症(EB,Epidermolysis bullosa)是一种罕见的遗传疾病,表现为皮肤非常脆弱,因日常的轻微摩擦而反复发作水疱。这种病不传染。大约5万个新生儿中会有一个患EB。所有的种族都会患EB,且男女比例相同。得此病的孩子患皮肤癌的风险非常高,并且没有任何已知的有效治疗方法。

大疱性表皮松解症是世界上最痛苦的疾病之一。患者的皮肤脆弱得犹如蝴蝶的翅膀,因此他们又被称为“蝴蝶宝贝”。好发于肢端及四肢关节伸侧,严重者可累及机体任何部位。皮损愈合后可形成瘢痕或粟丘疹,肢端反复发作的皮损可使指趾甲脱落。目前尚未找到治疗大疱性表皮松解症安全、有效且廉价的方法。及时处理破损皮肤,是提高患者生活质量的唯一办法。
大疱性表皮松解的治疗主要针对其继发感染,原则为精心护理,保护局部,避免外伤、摩擦受热防止继发感染。生活在凉快的环境中并避免高温对一些患者是有益的。因为在出生时或婴儿早期发病,大多由创伤引起,对患儿的护理是困难的例如避免引起创伤的活动。
爱丽丝梦游仙境症——视微症

爱丽丝梦游仙境症(Alice in Wonderland Syndrome,简称AIWS),又称“视微症”,属于一种罕见眼疾,是神经学上的一种高度迷惑性现象,以致影响到人类的视觉感知。其症候表现为:长时间观察一种事物,会突然像爱丽丝漫游仙境一样,周遭的事情忽然变大,或者忽然变小。病人有时候眼前会出现马赛克的视觉效果,有的患者则会出现时空扭曲感。

爱丽丝梦游仙境症候群,患者发病时意识都清醒,不会有昏睡、呕吐、激烈头痛、焦躁混乱等脑炎或脑膜炎般的症状;患者可以跟家人正常对话,但会有影像变形或视觉幻影的现象,如所看的人像会变小、变大、变远、变近、甚至变形。
依据我们的经验及某些文献上的记载,大都发病在夜间,有人推测可能黑夜较无法和周遭环境作相对大小比较之故。 因爱丽丝梦游仙境症候群的病程愈后,大都非常轻微,很多家长经历孩子一夜的辄疼后,就完全康复,根本没打算到大医院去求诊。因此,孩子感冒其间或感冒后,若有奇怪的言行举止,先不要怀疑小孩子是吃错药了,应和就诊的小儿科医师讨论此病的可能性。
早衰症

早衰症属遗传病,身体衰老的过程较正常快5至10倍,患者样貌像老人 ,器官亦很快衰退,造成生理机能下降。患此罕见疾病的儿童,即使只有16岁,但看上去好像六七十岁的老人。患病儿童一般只能活到7至20岁,大部分都会死于衰老疾病,如心血管病,现时未有有效的治疗方法,只靠药物针对治疗。
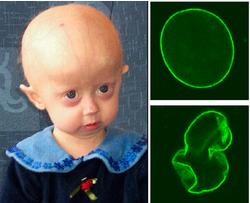
症病童较常出现的症状包括:脱发、较晚长牙、身材矮小及皮下脂肪减少等,但病童的心智年龄大多与同龄儿童无异。专家指出,病童大多死于心血管疾病等衰老病。目前早衰症的疗法目前没有一项被证实是有效的。大部分的治疗集中在减少并发症,例如冠状动脉绕道手术或低剂量阿司匹林。患者也可能受惠于高热量饮食疗法。
皇室病——血友病
血友病(Hemophilia)是一组由于血液中某些凝血因子的缺乏而导致患者产生严重凝血障碍的遗传性出血性疾病,男女均可发病,但绝大部分患者为男性。包括血友病A(甲)、血友病B(乙)和因子XI缺乏症(曾称血友病丙)。前两者为性连锁隐性遗传,后者为常染色体不完全隐性遗传。血友病在先天性出血性疾病中最为常见,出血是该病的主要临床表现。

19世纪,一个名为血友病的遗传性疾病因近亲结婚波及欧洲四个国家的皇室贵族,这在历史上绝无仅有。这种致命的疾病通过欧洲皇室的深重灾难向世人说明,人类基因遗传有其自身规律,了解、尊重这种规律是保护自身和亲人生命健康的第一要务。
出血是血友病的主要临床表现,患者终身有自发的、轻微损伤、手术后长时间的出血倾向,重型可在出生后既发病,轻者发病稍晚。血肿压迫神经可导致受压神经支配区域麻木、感觉丧失、剧痛肌肉萎缩等;舌、口腔底部、扁桃体咽后壁、前颈部出血,则可引起上呼吸道梗阻导致呼吸困难,甚至窒息而死,局部血管受压迫可引起组织坏死,血友病患者因凝血因子药物短缺而时常面临危险。
猫叫综合征
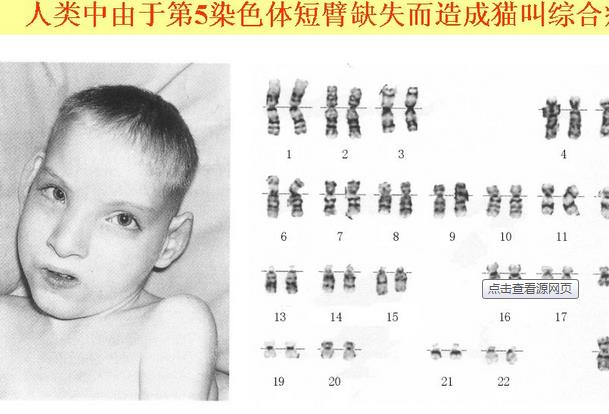
猫叫综合征,患者第5号染色体短臂缺失,故又名5p—综合征,为最常见的缺失综合征,因婴儿时有猫叫样啼哭而得名,其原因在于患儿的喉部发育不良或未分化所致,发病率约为1/50000,女患者多于男患者。猫叫综合征有猫叫样哭声并伴有特殊面容:头小,圆月脸不对称,呈惊恐状;眼距增宽,内眦赘皮,眼角下斜,斜视,白内障,视神经萎缩;鼻梁宽,小下颌,偶见唇腭裂,错咬合,耳位低,发育不良,颈短。新生儿期肌力低下,明显的智力低下、智力发育迟缓,如2岁后才能坐稳,4岁后才能独立走路,成人期后多动及破坏性行为。

近日,上海的一对年轻父母的孩子被确诊为猫叫综合征。11年了,孩子仅能说10个字以内短句,不能表示大小便,甚至对其他人攻击。这个11岁男孩的头比常人要小,眼睛间距比正常孩子宽。听不懂他在讲什么,但是从很远的地方就能听到一阵阵猫叫的喊声。他妈妈表示,刚出生时孩子吸吮能力很弱,不能自己喝奶。想尽了各种办法,最终用吸管将奶一点点滴进孩子嘴巴里。孩子42天检查的时候,医生发现孩子面部异常,害怕声音和光,更害怕陌生人。3岁时,染色体检查揭开了猫叫的秘密:“猫叫综合征”。该病全世界只有200例,我国仅20例左右,目前尚无治愈方法。
结语:
罕见病,又称“孤儿病”,是指盛行率低、少见的疾病,风靡全球的冰桶挑战让我们对罕见病有了进一步的了解,病罕见,关爱不能“罕见”,让我们持续关注罕见病,让关爱继续进行。